We have presented the following nine methods to predict RNA interface residues on a protein surface. The methods are based on propensity of residue occurrence in the interface of protein and RNA molecules observed in protein-RNA complex structures. Every method requires protein 3D structure coordinate. Some of them also requires multiple sequence alignment.
From a unique set of protein-RNA complex 3D structures, frequencies of
each residue occurrence can be calculated as;
 .
.
The first frequency is that of residue occurrence at the surface of a
protein, and the second frequency is that in the interface.
A singlet propensity for each residue is given by;
 .
.
When we consider a pair of residues in the interface and at the surface,
there could be some additional information. A pair residue is defined as
residues within 7A CB distance. The frequencies can be calculated as;
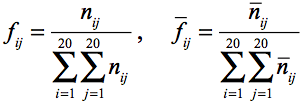 .
.
A pair frequency can also be expressed using singlet frequency;
 .
.
The coefficients C and D tell the effect of pairing.
Therefore, we define doublet propensity as;
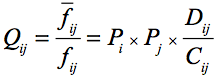 .
.
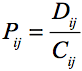 .
.
Functional information can also be found in comparison of homologous
sequences. We define a single residue profile at each site of the
alignment as;
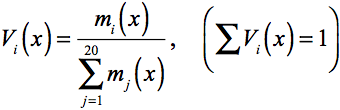 ,
,
and a double residue profile at two sites of the alignment as;
 .
.
Using the values given above, we can preform the following nine types of score calculation and predict interface residues;
- Multiple sequence profile score [P]:

- Singlet score [S]:

- Singlet plus profile score [SP]:

- Averaged singlet score [AS]:

The averaging of the score is performed amongst the residues within 7A distance from the residue in concern. - Averaged singlet plus profile score [ASP]:

- Singlet and doublet score [ASD]:

- Singlet and doublet plus profile score [ASPD]:

- Averaged singlet and doublet plus profile score [A2SPD]:

- Averaged singlet and doublet score [A2SD]:

A jackknife benchmark test of the methods tells that doublet propensity
and profile contain significant amount of information.

Use PDB format file for protein 3D structure coordinate data. The format
looks like the following;


For the prediction, HEADER, TITLE, ATOM and END rows are required. Other rows are not necessary.
Be sure to change MSE to MET, SEC to CYS, ... and those rows should be ATOM (not HETATM), so that all the protein residues are described in ATOM rows and residues in the ATOM rows are standard amino acids.
For the detail of the format, please visit wwPDB
For the methods that require profile, a multiple sequence alignment
should be given by ClustalW format;

The first line should start with "CLUSTAL W" and two blank lines should follow it. An alignment is blocked with 60 positions, and each block is separated by two lines, one of which contains marks for indicating conservation of residues and the other of which is a blank. In each block, an amino acid sequence start at 19th column.
This input is to specify an amino acid sequence in the alignment that is exactly the same with the one of PDB file. If there is a discrepancy in residue name between PDB file and the sequence specified by ID, then the prediction does not work.